
近日,我所催化基础国家重点实验室生物无机催化研究组(507组)叶生发研究员团队与台湾师范大学李位仁教授和马克思-普朗克化学能源转化研究所Schnegg教授合作,在研究金属酶及模拟配合物催化分子氧活化的反应机理中取得新进展。
氧化反应通常是放热反应,具有很高的驱动力,但需要克服氧分子从自旋三重态转换到自旋单重态产生的高动力学势垒。自然界中存在一系列金属酶,能够高效地活化分子氧进而将底物功能化,即使在室温条件下该反应也表现出很高的效率。目前被广泛接受的反应历程是分子氧与金属酶的活性位点结合,使其进行单电子还原形成金属超氧配合物中间体,并逐步还原为金属过氧配合物、金属端基氧配合物。尽管过去已有报道,如何实现金属超氧配合物到过氧配合物的转化在很大程度上仍然是未知的,这增加了对分子氧活化的反应机理的研究难度。
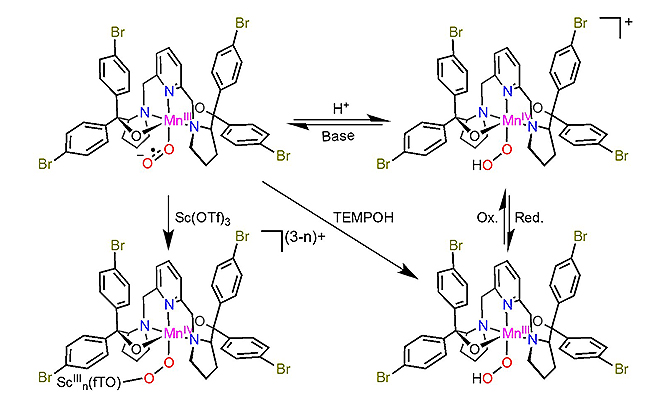
叶生发团队与其合作者早期研究发现,二价锰、铁和钴与分子氧的反应生成三价金属超氧配合物,该转瞬中间体可通过氢原子攫取(HAA)转化为三价金属过氧化氢配合物(J. Am. Chem. Soc. 2016, 138, 8862-8874; J. Am. Chem. Soc. 2016, 138, 14186-14189; Inorg. Chem. 2019, 58, 9756-9765.)。近期,该团队采用共振拉曼、多频率EPR、理论计算等手段研究了三价锰超氧配合物与Bronsted酸和Lewis酸的反应。结果表明三价锰超氧配合物经过质子或金属耦合的电子转移分别转化为四价锰过氧化氢配合物和Mn(IV)/Sc(III)过氧配合物,由此证实了金属超氧配合物的两亲性。本项工作首次实现了利用质子或金属耦合的电子转移将金属超氧配合物转化为过氧配合物,为理解金属酶和人工催化剂的分子氧活化和转化机理提供新思路。
该研究成果发表在《美国化学会志》(J. Am. Chem. Soc.)上。该工作得到中科院专项人才计划A类项目的资助。(文/图 姜洋)


